Molecular glue degraders (MGDs) are small molecules that induce or stabilise protein–protein interactions, often by recruiting an E3 ligase to a target protein and triggering its ubiquitination and degradation. They are emerging as an important class within targeted protein degradation (TPD) because they can access targets that are difficult to modulate with conventional small molecules. This article explains what molecular glue degraders are, how they work, how they differ from PROTACs, and what innovators should consider when building an IP strategy around MGDs.
How are Molecular Glues Different from PROTACs?
This is the second instalment of our commentary on targeted protein degradation (TPD) (read our earlier blog on PROTACs here). Unlike PROTACs, which are typically modular bifunctional molecules with separable ligand components, molecular glue degraders rely on a more integrated architecture in which the degrader reshapes or stabilises an interaction surface. This makes MGD design less amenable to “platform” approaches and has important consequences for patent drafting, and freedom-to-operate analysis.
What is a Molecular Glue?
Molecular glues are small, often ‘conventional‑looking’ molecules that rewire protein–protein interactions (PPIs) that would otherwise be weak or absent in cells. Put simply, they ‘stick’ two proteins together by bridging them directly or by reshaping one protein surface to make the interaction with the second protein favourable.
Molecular glues are not new. The modality was first identified in the early 1990s. What is new is the field’s shift from retrospective mechanism‑spotting years after regulatory approval towards actively designing molecular glue behaviour from the outset. This shift has obvious implications for drug discovery and brings IP considerations and pitfalls that are easy to overlook if MGDs are treated as just another small‑molecule programme.
The transition from serendipity to strategy is set to be marked by iberdomide (CC-220); one of three MGDs developed by BMS/Celgene in phase III clinical trials. BMS/Celgene have a rich history in bringing molecular glues to the clinic, so it is no surprise that iberdomide, which has a PDUFA date of 17 August 2026 (the date by which the US FDA must review and issue a decision on BMS’ new drug application), might become the first drug to receive approval with a prospectively understood and validated MGD mechanism.

How do Molecular Glues Work?
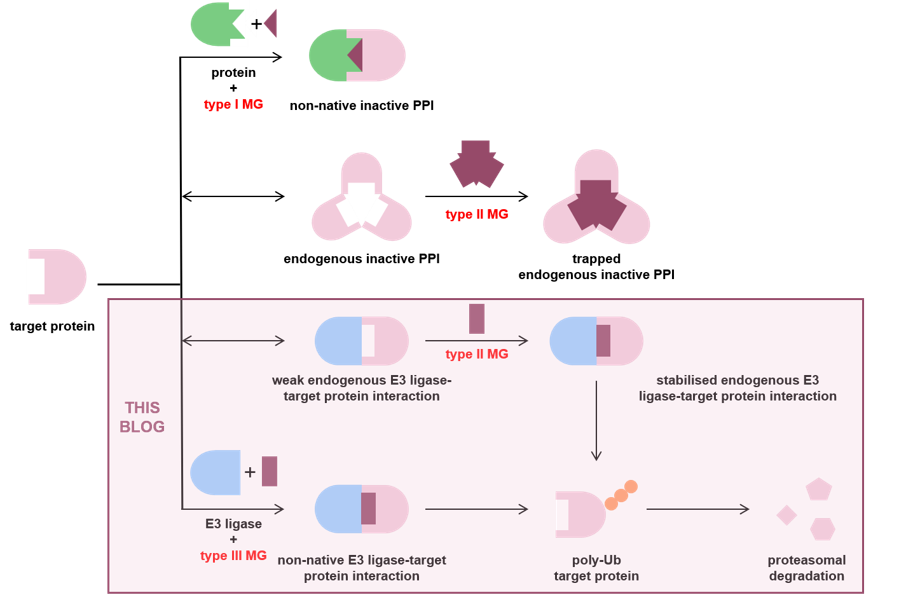
Although this blog focuses on molecular glue degraders (MGDs), molecular glues are not limited to degraders. A published classification (based on clinically approved molecular glues) groups them into three broad functional types summarised graphically below.
-
Type I molecular glues induce inactive non-native PPIs to indirectly inhibit a harmful endogenous PPI;
-
Type II molecular glues stabilise or trap endogenous PPIs which may be active or inactive (examples of both are shown in the figure below); and
-
Type III molecular glues induce active non-native PPIs, which can have different outcomes including ubiquitination and degradation.
Molecular Glue Degrader Architecture
MGDs contain an E3 ligase-recruiting component that anchors the molecule to a specific ligase (typically CRBN), together with a substrate-recognition component that reshapes the ligase surface and enables formation of a ternary complex between the ligase, the glue and the target protein. This can lead to ubiquitination and proteasomal degradation.
Although MGDs are still functionally bifunctional, they are structurally distinct from PROTACs. In MGDs, the substrate-recognition component is closely adapted to the local environment of the recruited E3 ligase. As a result, MGD chemistry is generally less modular, making broad “platform” filing strategies harder to sustain.
Molecular Glue Degrader Landscape: Clinical Pipeline and Key Targets
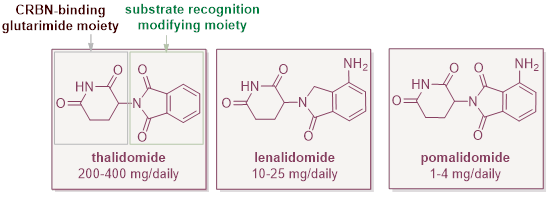
Historically, several of the best known MGDs (thalidomide, lenalidomide and pomalidomide – shown below) entered the clinic without any intent to discover a degrader. Their glue mechanism was only elucidated after clinical activity was already established. This is why molecular glues have so far carried a reputation for serendipity and have not typically featured in the stated drug development aims of most pharmaceutical companies.
First-Generation Molecular Glue Degraders
That picture is now changing, and there is a growing pipeline of clinical candidates that have resulted from purposeful investigation and exploitation of the MGD mechanism. This next generation of MGDs, summarised in the table below, is also moving beyond the historic confines of haematological indications and IKZF1/3 targeting.
|
Designation (Compound) |
Developer/ Sponsor |
Degraded Target(s) |
Phase (Status) |
Therapeutic Indication(s) |
|
Iberdomide (CC‑220) |
Celgene/BMS |
IKZF1/3 |
Phase III |
RRMM; SLE; NHL |
|
Mezigdomide (CC‑92480) |
Celgene/BMS |
IKZF1/3 |
Phase III |
MM; RRMM |
|
Golcadomide (CC‑99282) |
Celgene/BMS |
IKZF1/3 |
Phase III |
HR-LBCL |
|
CC‑91633 (BMS‑986397) |
Celgene/BMS |
CK1α |
Phase I |
RRAML; RRMDS |
|
E7820 (mysidamide) |
Eisai |
RBM39 |
Phase II |
RRAML, MDS, CMML |
|
MRT‑2359 |
Monte Rosa Therapeutics |
GSPT1 |
Phase I/II |
MYC-driven solid tumours |
|
MRT‑6160 (partnered) |
Monte Rosa / Novartis |
VAV1 |
Phase I |
Autoimmune disease |
|
NVP‑DKY709 |
Novartis |
IKZF2 |
Phase I |
Advanced cancers (immune modulation; NSCLC, melanoma) |
|
NEO-811 |
Neomorph |
ARNT (HIF-1β) |
Phase I/II |
locally advanced or metastatic non-resectable ccRCC |
|
CFT‑7455 |
C4 Therapeutics |
IKZF1/3 |
Phase I/II |
RRMM; NHL |
|
ICP‑490 (structure not yet known) |
InnoCare |
IKZF1/3 |
Phase I/II |
RRMM; NHL |
|
GT‑919 (structure not yet known) |
Gluetacs Therapeutics |
IKZF1/3 |
Phase I |
RRMM; RRNHL |
|
BTX‑1188 |
Biotheryx |
IKZF1/3 |
Phase I |
Advanced hematologic & solid malignancies |
|
ST-01156 (structure not yet known) |
SEED Therapeutics |
RBM39 |
Phase I |
Ewing sarcoma, hepatocellular carcinoma and other related tumors |
In principle, any of the hundreds of E3 ligases could be recruited by molecular glue degraders; in practice the E3 ligase of choice is Cereblon (CRBN) because it has a well-understood binding pocket suitable for anchoring molecular glues and modifying CRBN’s substrate recognition surface. Only E7820 and ST-01156 (in bold above and E7820 circled below) do not make use of the glutarimide moiety. Notably, IKZF1/3 are also the prevalent degradation target.
These common choices come with a predictable consequence: the same ‘anchor’ chemistry (a glutarimide moiety) and substrate-recognition modifying motif (an isoindolinone moiety) recur across many programmes (depicted below). This creates a congested IP landscape compared to that facing classic small molecule drugs.
Next-Generation Molecular Glue Degraders

Patent Strategy for Molecular Glue Degraders: Claim Scope, Data and Freedom to Operate
Patent strategy for MGDs requires a different approach from conventional small molecules. Although MGDs may look like ordinary small molecules, their mechanism, crowded prior art and dependence on functional data create distinct issues for claim breadth, plausibility, inventive step, amendment strategy and freedom to operate.
Foremost among these considerations are scope and evidence. MGD applications are the target of more attacks against claim breadth than other small molecule drugs, especially any parts of the scope that are not supported by data. This is in part because MGDs operate by a multi-step pathway as opposed to the one-step occupancy-driven mechanism of conventional small molecules, so Patent Offices may not be satisfied by data for a single mechanistic step. Examiners may argue that even small structural changes can abolish glue-like behaviour, even if CRBN binding is retained. Innovators can expect Patent Offices to push for increasingly narrower composition of matter (COM) claims and to require more justification for the extent of generalisation.
To avoid these pitfalls, data for MGD applications should ideally go beyond CRBN binding affinity, for example by also validating ternary complex formation, ubiquitination and degradation. They should also include functional language, which can help to bridge the evidential gap for exemplified compounds which do not have data for all these downstream steps. The broadest scope will only be achievable by a combination of robust data sets and precise functional language.
The crowded prior art may also affect the assessment of inventive step. For example, at the EPO novel MGDs may be assessed as selection inventions if they are encompassed by or overlap with earlier genus claims. This requires proving a credible technical advantage over the prior art – a distinction that will need to be underpinned by comparative data. If the genus in the prior art is too broad it might also be argued in line with the point above that the excessive scope is not enabled to sidestep the higher inventive step requirements for a selection invention
Amendment strategy is another area that rewards early planning. The EPO’s strict assessment of added subject-matter during amendment can present a serious obstacle unless amendment strategy is given detailed thought during the drafting of the application. This is especially true of MGDs, where the crowded prior art landscape may require precise amendment to carve out earlier disclosures without sacrificing more claim scope than is necessary. Layered, explicit and combinable fall-back positions will play an important role in obtaining the broadest possible protection for MGDs.
The wider filing strategy must also take into account all the above considerations. Unlike some classical small‑molecule programmes, MGDs often do not benefit from early, more-speculative filings. Broad early disclosures lacking convincing data may struggle on plausibility and inventive step, while simultaneously impeding subsequent, more focused filings. In an EPO context, MGD portfolios benefit from fewer, more focused filings arising from maturer programmes centred on validated leads with demonstrated degradation. These filings can then be followed by carefully sequenced secondary applications once the degradation biology and SAR are better understood.
This approach trades some breadth for stronger, more defensible claims – often a worthwhile exchange in the crowded MGD space.
Finally, freedom to operate also requires its own particular care. Although MGDs lack the modular architecture of PROTACs, they often sit squarely on top of a well‑established chemical space, particularly around E3 ligase binders, and one in which functional language is widely used. A MGD programme may therefore intersect with prior rights from multiple fields, with the risk not always apparent from structural comparison alone. Early, iterative, and carefully constructed FTO assessment is therefore essential.
Future Outlook for Molecular Glue Degraders
MGDs are set to take on a more significant role within the broader TPD landscape. Their relatively small size brings clear advantages, both in terms of Lipinski-compliant properties and ease of manufacture compared with other degrader modalities. In addition, their mechanism can deliver therapeutic outcomes that are difficult or even presently impossible to reach via classical occupancy‑driven inhibition and offers promising candidates for combination with delivery platforms.
It will be interesting to see how the space evolves beyond the well-trodden CRBN chemistry as new E3 ligase binders are developed, and if any families of E3 ligases show common binding or surface-modification behaviour to permit so-called ‘platform filings’ becoming part of the MGD IP. Steps in this direction are already being taken, not least by Eisai’s E7820 and SEED Therapeutic’s ST-01156 (see above), which bind DCAF15. Further exciting work is being done by Amphista Therapeutics whose pre-clinical MGDs bind DCAF16, FBXO22, and DCAF11.
For now, the take-home message is that securing the best protection for MGDs requires a carefully considered approach distinct from other similarly sized small molecule therapeutics.
This blog was co-authored by Tim Nash and Andrew Pitts.


.png)
.png)