The GLP-1 landscape changed on 1 April 2026, when the FDA approved Eli Lilly's orforglipron under the brand name Foundayo as the first non-peptide, small-molecule GLP-1 receptor agonist (GLP-1RA) anywhere in the world. The approval matters because it validates a long-sought oral small-molecule route to GLP-1 agonism, with potentially major consequences for convenience, manufacturing scale, pricing, and patent strategy.
For two decades, GLP-1 drugs have been an exclusively peptide story. Exenatide, a natural peptide isolated from the saliva of the Gila monster, launched the field in 2005. After modifications in the lab, liraglutide, semaglutide and tirzepatide turned it into the most commercially significant new drug class in a generation. Each peptide is manufactured by solid-phase synthesis or recombinant fermentation and is dosed predominantly by injection, because oral bioavailability is only around 1% and oral administration also carries fasting requirements.
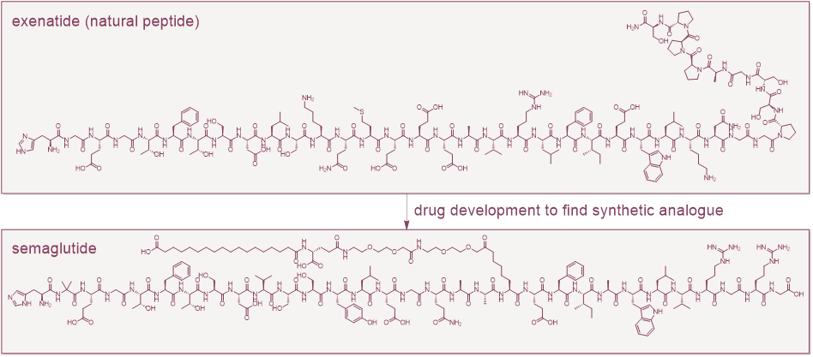
This post examines why the approval matters, how small-molecule GLP-1 agonists compare with peptides, and what the shift means for patent strategy and the pipeline ahead.
Why was GLP-1 receptor agonism more challenging for small molecules?
The GLP-1 receptor is a Class B G protein-coupled receptor. Endogenous GLP-1 is a 30-residue peptide that engages the receptor's large extracellular domain and then threads its N-terminus deep into the transmembrane bundle. It is a famously hard target for small-molecule agonism. Allosteric (off active site) activators were reported in the literature as early as the mid-2000s, but they had poor potency, poor pharmacokinetics, species-selective activity (human but not rodent), and none made it close to the clinic.
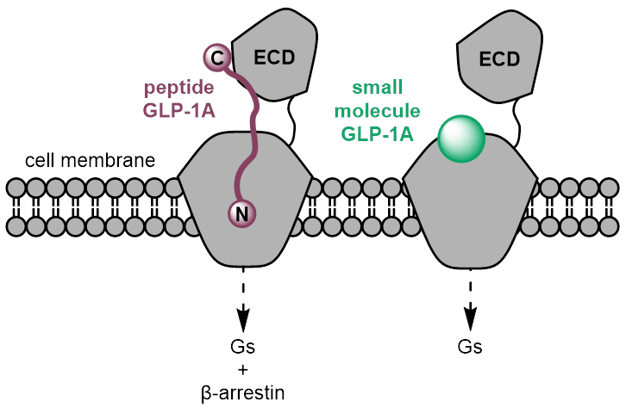
The critical breakthrough was structural. Cryo-EM structures of peptide activated GLP-1R published from 2017 onwards (see Zhang et al and Liang et al) provided the first high-resolution view of class B GPCR activation. In 2020 the first small-molecule structure (Zhao et al) revealed a druggable transmembrane sub-pocket distinct from the peptide-binding interface. Later that same year the orforglipron structure (Kawai et al) showed the now-recognised Trp33 ECL1 anchor that underlies the human-receptor selectivity of the modern small-molecule series. Compounds binding there can stabilise the active conformation without needing to mimic the full peptide-extracellular-domain interaction. Most of the small molecules now in development bind in this same general region and exploit the Trp33 anchor.
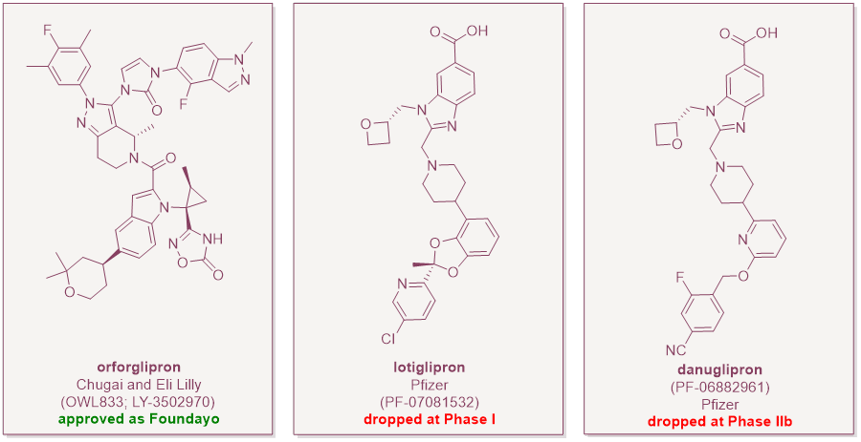
The success of orforglipron is even more impressive considering that some competitor small-molecule GLP-1RAs have not progressed. The field is so far proving to be unpredictable. Pfizer’s lotiglipron was scrapped in June 2023 over transaminase elevations in Phase I studies. Danuglipron made it further to Phase IIb, but Pfizer ended the entire program in April 2025 after a single asymptomatic case of potential drug-induced liver injury and dropped its third oral candidate a few months later. Hepatic safety is now a class consideration.
What does Foundayo's approval mean for patients?
The FDA cleared orforglipron for chronic weight management in adults with obesity, or overweight with at least one weight-related comorbidity, based on the ATTAIN clinical program which showed placebo-adjusted weight loss of 7.5% to 11.2% in non-diabetics (ATTAIN-1) and 5.1% to 9.6% in diabetics at different doses, along with the expected glycaemic benefits (ATTAIN-2).
Two points of the regulatory authorisation of orforglipron are worth noting. First, the approval came a mere 50 days from filing thanks to the FDA's new Commissioner's National Priority Voucher pilot, reportedly the fastest NME clearance since 2002. Second, the labelling is genuinely different to the peptides: unlike the oral semaglutide pills, orforglipron can be taken at any time of day, with or without food or water. That is the practical payoff of a real small molecule; bioavailability that doesn't depend on a specific gastric environment.
How do the small molecule GLP-1RAs compare to the peptides?
On mechanism, the peptides and the small molecules end up at the same destination (Gs activation, cAMP elevation, the canonical incretin pharmacology) but they get there via different binding modes and with different signalling profiles. The peptides occupy the extracellular domain and the orthosteric pocket, whereas the small molecules wedge into the transmembrane region around Trp33. Several of the small molecules (e.g. aleniglipron and orforglipron) are functionally Gs-biased and recruit β-arrestin poorly (which is typically involved in turning off the GLP-1 receptor's signal by promoting internalization and downregulation and mediating nausea and vomiting). The theoretical benefit compared to the peptides is reduced receptor desensitisation and a different tolerability profile, though whether that translates to clinically meaningful differences in nausea, vomiting and discontinuation rates is still under investigation.
On efficacy, orforglipron sits between oral semaglutide and injected tirzepatide. In the ACHIEVE-3 head-to-head against oral semaglutide in T2D, 36 mg orforglipron produced about 9.2% body weight reduction versus about 5.3% on 14 mg oral semaglutide, and superior HbA1c reduction. Against injectable tirzepatide, orforglipron loses. A network meta-analyses put tirzepatide clearly ahead on weight loss, which is the expected outcome for a dual GLP-1/GIP agonist versus a single GLP-1 agonist. In ATTAIN-MAINTAIN, patients who switched from semaglutide or tirzepatide to orforglipron broadly maintained their losses, which is a sensible secondary commercial position; a maintenance pill to be used after concluding the main weight loss phase with an injectable.
On safety, the GI profile broadly mirrors the peptides. The cloud hanging over the entire class right now is hepatotoxicity. Danuglipron and lotiglipron both fell to liver signals, and Pfizer's experience has made the FDA and other agencies acutely sensitive to transaminase data in oral GLP-1 programs. Orforglipron's filing did not flag a hepatic signal, but the long-term real-world data set is just beginning.
On manufacturing, this is where the small molecules really change the economics. A peptide GLP-1 needs solid-phase synthesis (semaglutide is a 31-mer with non-natural modifications and a lipid chain) or recombinant expression followed by complex modification. In comparison, a small molecule like orforglipron is a roughly 16-step convergent organic synthesis from common starting materials (see, e.g., Part I and Part II of a key intermediate synthesis from the Lilly process team). The supply chain is shorter, the cost-of-goods is lower, and the global capacity is essentially uncapped. Contrast this with the peptides which faced major supply issues between 2023-2025 that compromised diabetic patient safety and led to higher prices, unequal access and a rise in fake medicines. For low- and middle-income markets this all matters enormously.
A different game from the peptides – what are the patent considerations?
The transition to small molecules is consequential. The patent strategy for a peptide GLP-1RA is in some respects quite different from the strategy for a small molecule, and orforglipron has already become an instructive case study.
Small-molecule composition of matter (COM) patents feature a classical generic Markush drawn around examples with the lead clinical candidate(s) at the core. By contrast, peptide GLP-1RA COM patents are built on a different logic: claims that cover the precise peptide sequence and its specific modifications, with limited genus scope because the SAR around peptide modifications is exquisitely position-specific. Exclusivity on the peptides has proven robust because the claim protects the key peptide and, even though the generic scope may be limited, there is little structural room for a successful follow-on outside the claim. Small molecules have a much larger serviceable chemical space which competitors will try to occupy if it is not covered by the generic Markush formula.
Unlike peptides, a small molecule sometimes provides the possibility of one or more solid forms. For the peptides the lifecycle opportunities mainly relate to formulation, conjugation, and the injection device. Orforglipron and its small-molecule successors open the door to crystalline form patents as well as process/synthesis, key intermediate, oral formulation, prodrug, and stereochemistry patents. The small-molecule GLP-1RA estate will have many more lifecycle-extension opportunities for the originator.
Finally, peptide proprietors benefit from the fact that regulatory and market entry barriers are not trivial. The fast-paced Indian and Chinese semaglutide programs notwithstanding, US and EU regulatory entry in particular carries real cost and higher synthetic complexity. Peptides sit somewhere between small-molecules and biosimilars in this regard. Unlike small-molecules, generic peptides often require a clinical efficacy trial, an immunogenicity assessment, and impurity determinations. Costs can be in the single millions for small molecules compared to mid- to high tens of millions for peptides. The eventual cost-of-entry collapse will be much sharper for the small molecules than what is coming for the peptides. This makes the strength of the small molecule patent estates even more critical to retain hard-won market share after loss of the COM patent.
Outlook - what next?
There are three likely waves in the near future for small-molecule GLP-1RAs.
The first wave will hopefully be the approval of more small-molecule GLP-1RAs. Aleniglipron (Structure Therapeutics) is heading into Phase 3 in mid-2026. ECC5004/AZD5004 (AstraZeneca/Eccogene) and several Chinese programs are not far behind (see, e.g., here and here). Within three to four years, the oral small-molecule sub-class is likely to have at least one or two more approved members, and price competition will start to put pressure on the injectables in cash-pay and lower-income markets.
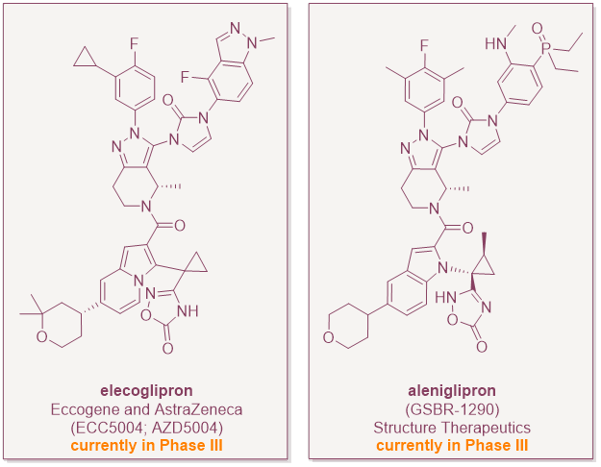
The second wave will be the invention of combinations and next-generation pharmacology. Structure Therapeutics has already disclosed an oral small-molecule amylin program (ACCG-2671, ACCG-3535) intended for fixed-dose combination with GLP-1RAs, and is working on small-molecule agonists at GIP, glucagon and apelin receptors. The peptide world has been racing toward dual and triple agonism (e.g. tirzepatide and retatrutide, respectively); the small-molecule world will look to do the same, but likely as fixed-dose combinations of two or three orals rather than as single multi-functional peptides until true dual and triple small-molecule agonists can be found. It will be difficult to engineer balanced agonism into a single small-molecule because the sequence hybridisation of the dual and triple peptide agonists has no obvious chemical analogue. This has knock-on implications for patent filings and life-cycle management of the small-molecules in this space.
The third wave, which is perhaps more of an open question than the first two, is whether hepatic safety can be reliably designed out. Pfizer's experience suggests it is a chemotype-dependent problem, not a class problem. Orforglipron's lack of a liver signal provides hope that there is a chemical series that avoids it. But the field will watch transaminases on every new candidate for years. If a second drug-induced liver injury signal emerges post-marketing on any approved small-molecule GLP-1, the regulatory and commercial framework will recalibrate sharply.
For patent practitioners in this field, the lesson is that this class is shifting from a peptide paradigm to a small-molecule paradigm. Small-molecule GLP-1RA litigation over the coming decade will likely generate more numerous, more independently contested patent challenges than peptide GLP-1RA litigation has. These challenges are primed to look chemically and procedurally like what the field has previously seen in oncology around the CDK4/6 cluster (palbociclib, ribociclib, abemaciclib) and in the SGLT2 inhibitor space. Lessons from these litigation stories should be drawn upon now to draft strong patent applications that will weather the inevitable future challenges.

.png)
